Best practices for single-cell analysis across modalities
跨模式单细胞分析的最佳实践,于 2023 年 3 月 31 日发表于 nature reviews genetics。最近在研究单细胞分析相关的资料,因此记下这篇笔记。文章中包含了 Transcriptome、Chromatin accessibility、Surface protein expression、空间转录组等多个方面,但就近期关注点而言,只精简记录单细胞转录组部分的笔记。如果有需要,可以阅读原文。
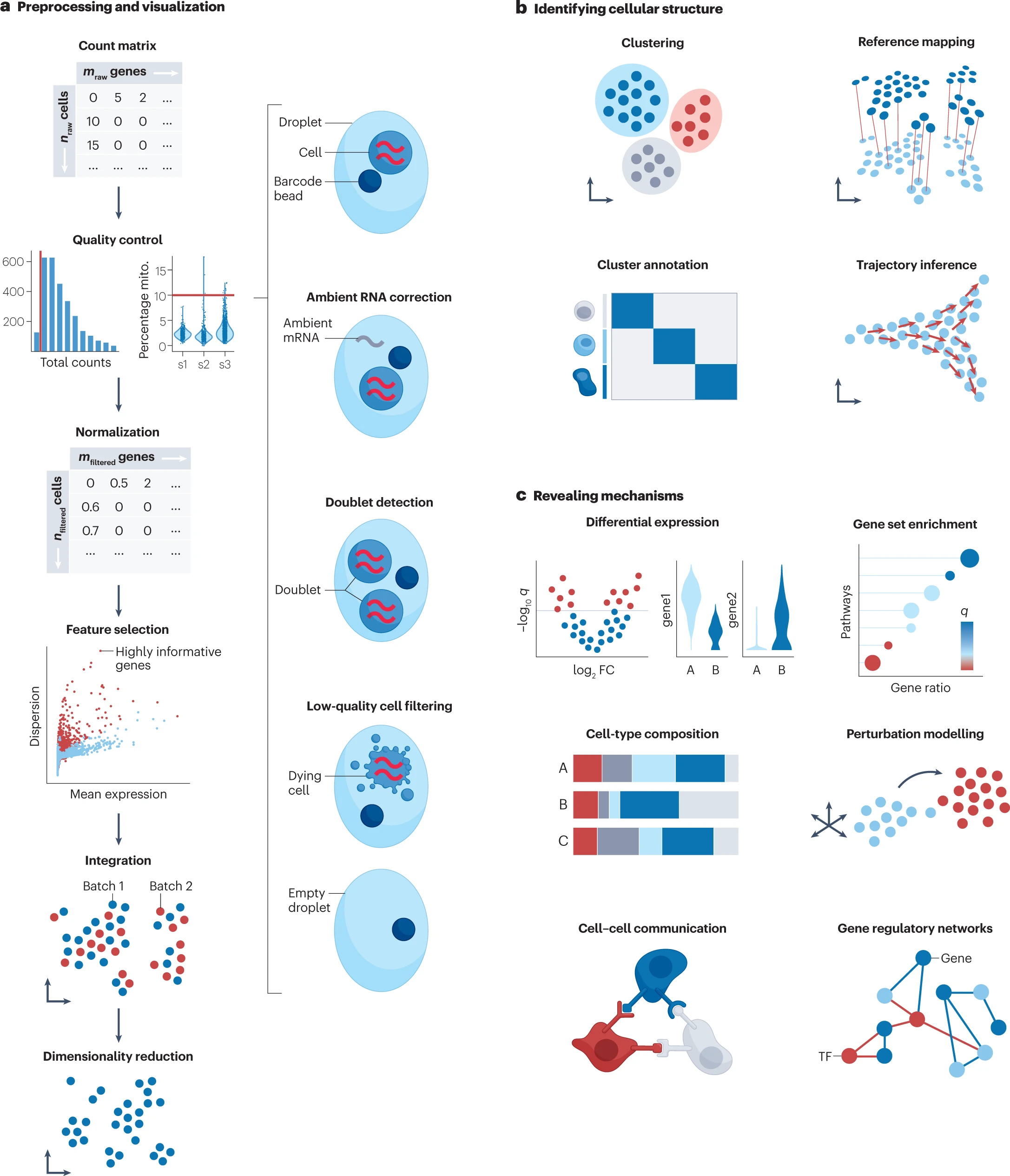
从原始计数矩阵到高质量单细胞数据
原始计数矩阵是直接从测序数据定量得来的矩阵,一般根据每个细胞 barcode 与 UMI 确定细胞中各种 mRNA 的数量,然而原始数据中包含着各种系统噪声和误差(比如来自建库操作、样本质量、测序批次等),因此在这一预处理过程中试图去除误差。这一过程涉及到质量控制、归一化、数据矫正和特征选择等过程,还可能会涉及到消除批次效应以整合多批次数据的过程。
滤低质量细胞和噪声校正 Filtering low-quality cells and noise correction
大多数分析过程假定每个单细胞液滴中都包含来自单个完整细胞的 RNA。细胞质量差(比如死细胞)、溶液中破裂细胞产生的 RNA 污染、同时捕获多个细胞的情况违反这一假设,因此需要在进一步分析之前去除这部分数据。
-
低质量细胞:检测基因数量过少、深度过低或是线粒体计数(植物还有叶绿体)过多的细胞都被认为是低质量细胞。一般通过手动设置的阈值来进行识别和过滤,可以参照先前文章或是基于中值绝对偏差数对样本进行过滤。需要综合考虑过滤的指标,同时注意质量控制是在样本水平上进行的,因为样本之间的阈值可能会有很大差异。
-
中值绝对偏差数(Median Absolute Deviation, MAD)
基于中值绝对偏差数(Median Absolute Deviation, MAD)的样本自动过滤是一种统计方法,用于识别和剔除数据集中的离群值。这种方法主要有以下几个步骤:
- 计算数据集的中位数(median)。
- 对每个数据点,计算其与中位数的偏差绝对值。
- 计算这些绝对偏差的中位数,得到MAD值。
- 选择一个阈值,通常是MAD的某个倍数(例如3倍MAD),用于判断数据点是否为离群值。
- 剔除大于阈值的数据点,即被认为是离群值的数据点。
这种基于MAD的方法对于处理非正态分布的数据集具有较好的鲁棒性,而且相对标准差受异常值影响更小,因此可以有效地识别和过滤异常值。
-
-
无细胞游离 RNA:通常存在于溶液中,并在建库过程中分配给每个细胞,产生一种类似背景值的效果。如果是细胞类群特异性标志物的 RNA 污染了其他细胞,后续分析中可能将不同的细胞群混合在一起。流行的 SoupX 等方法根据样本中「空液滴」的表达情况去除污染;CellBender 将环境 RNA 的去除制定为无监督贝叶斯模型,该模型不需要细胞类型特异性基因表达谱的先验知识。应当尽量不忽略去除环境 RNA 这一步,以提升下游分析的质量。
-
空液滴和双联体液滴(包含两个细胞的液滴):由不同细胞类型形成的双联体很难注释,并可能导致细胞类型标注错误。常见的双峰检测方法随机抽取两个细胞组合成模拟双联体,并将其与其他细胞测量结果进行比较。scDblFinder 采用此方法, 并在多项测试中准确性和计算效率优于其他方法。还可以应用多种不同检测方法并比较结果以提高准确性。
归一化和方差稳定化 Normalization and variance stabilization
不同细胞的 mRNA 含量不同,加上测序过程随机误差,将导致细胞有不同的初始计数。归一化可以矫正测序深度的差异,使不同细胞具有可比性。随后稳定方差以确保少数离群值对整体数据影响不会太大。
shifted logarithm transformation:使用缩放系数s的 log(𝑦/𝑠+1) 表现良好,但不应使用 CPM(counts per million)作为输入。
should not be used with counts per million as an input, as it reflects an unrealistically large overdispersion. 原文如此,但没看懂为什么,待补充。
通过使用一个公因数缩放所有基因,会假定由于细胞大小导致的测序深度差异可以忽略不计。然而对于异质性较大的数据集,其中包含了特性不同的各种细胞,其本身基因表达差异就较大,难以对每个基因找到一个合适所有细胞的系数。
此外还有 Scran 标准化和 Pearson 残差方法。shifted logarithm transformation 被证明可以更好地稳定方差以进行后续降维,Scran 在批量校正任务中表现良好,Pearson 残差更适合生物变异基因的选择和稀有细胞身份的识别。
消除混杂的变异来源 Removing confounding sources of variation
混杂变异可以来源于技术和生物,并且应该分开处理。
- 批次效应:多个样本的数据集可能会由于技术产生的批次效应,需要恰当的数据整合方法。canonical correlation analysis 与 Harmony 等线性嵌入模型在处理不同批次的简单整合上表现良好;在复杂任务重, atlas、scANVI、scVI、scGen等深度学习方法,及 Scanorama 等显性嵌入模型表现较好。scIB 包可用于使用上述基准的评估指标来评估整合效果。
- 生物因素:细胞周期等因素同样会引入误差,使得细胞间差异来源于细胞周期而非细胞类型。可以使用 Scanpy 或 Seurat 中内置的细胞周期标记与校正函数作为基础;Tricycle 等更复杂的方法对细胞异质性高的数据集表现良好。
选择信息特征和降维 Selecting informative features and reducing dimensionality
理想情况下,特征选择方法应优先选择在亚群间(而非亚群内)变化的基因,并且不影响亚群内更小亚群的可识别性。
通过拟合基因模型获得 deviance 来识别信息量大的基因,模型假定所有细胞基因表达一致,然后检验哪些基因违反假设,从而选择基因。此外,按此方法利用了原始计数对基因进行排序,不依赖归一化,不会因为数值中零值过多产生误差(zero inflation)。选择特征后可以通过 PCA 等方式进一步降低数据集维度,同时 PCA 结果可以进一步作为输入应用 t-SNE、UMAP、PHATE 等可视化降维方法。注意,最近有研究表明,仅仅依靠2D嵌入可能会导致对细胞之间关系的错误解读。因此,结果不应仅基于对结果的视觉检查来制定,而应结合其他定量评估。
为细胞集群确定亚群身份 From clusters to cell identities
从单个细胞到集群 From single cells to clusters
识别细胞群的第一步是将表达模式相似的细胞聚类成组,推荐使用 Leiden 算法进行聚类。
为细胞集群确定细胞身份 Mapping cell clusters to cell identities
建议分三步进行:自动注释 → 手动注释 → 验证。
- 自动注释:分为基于分类器的方法和基于参考数据集的映射方法。分类器可以参考 CellTypist 和 Clustifyr,两者利用先前注释过的数据集进行训练并参考大量基因进行分类;映射方法有 scArches、Symphony 或 Azimuth。这两类方法都依赖于参考数据的质量+模型质量+参考数据集对新数据的适用性。
- 手动注释:利用细胞群的特异性标志基因进行注释。
- 验证:需要仔细设计验证试验,并且对于复杂数据或稀有亚群,可能没有资料可供参考。
伪时间分析——从离散状态到连续过程 From discrete states to continuous processes
根据细胞表达模式的相似性,沿着轨迹对细胞进行排序的模型被称为轨迹推断或伪时间分析。这类分析的效果取决于数据集中存在的轨迹类型,Slingshot 对简单结构表现良好,PAGA 和 RaceID/StemID 对复杂结构表现更好,建议使用 dynguidelines 选择适用的方法。当预期结构未知时,应采用不同假设通过多种方法确认轨迹和假设。推断出的轨迹不一定有生物学意义。
利用 RNA velocity (mRNA 剪切、未剪切、降解的比例),比如考虑模型剪接动力学的 velocyto 和 scVelo,可以根据未剪接和剪接片段确定 RNA velocity:如果一个基因被激活了,未剪切的 RNA 会先于剪切的 RNA 产生。获得的 RNA velocity 输入 CellRank 来估计细胞命运。
谱系追踪根据细胞中观察到的变异(例如自然发生的基因突变)来推断其谱系模型,判断克隆群体中的细胞分裂历史,可以使用 Cassiopeia 提供的多种算法进行分析,建议对比多种算法的结果。同时还有能处理更加复杂谱系的 LineageOT 工具,以及追踪静态条形码的 CoSpar。
揭示机制 Revealing mechanisms
对数据进行注释后,可选的分析变得多样,根据研究问题选择要进行的后续分析。
差异基因表达分析 Differential gene expression analysis
目前单细胞的差异表达分析从两个思路进行:
- 样本层面的方法:将单个样本中的多个细胞细胞读数合并,生成人工合成的批量样本,再利用常规的转录组差异表达分析工具,比如 edgeR、DESeq2 或是 limma。
- 细胞层面的方法:利用广义混合效应模型(比如 MAST),对单个细胞进行建模。
各个工具之间的一致性和稳健性较低,但相对而言,为常规 DEG 设计的工具表现良好,而单细胞特异性的方法通常会低估基因表达的变异程度,因此倾向于将高表达的基因错误标记为差异表达基因。因此推荐 limma、edgeR 或 DESeq2。
基因集富集分析 Gene set enrichment analysis
常见的数据库包括 MSigDB、Gene Ontology、KEGG 与 Reactome 等,此外还有用于癌症信号通路的 PROGENy 和转录因子的 DoRothEA。一般采用超几何分布检验、GSEA 或 GSVA 进行富集分析。研究发现富集分析对基因集的选择敏感性大于统计方法,因此应当谨慎选择数据库。decoupleR 等框架提供了多个不同数据库的访问方法。为常规转录组开发的方法可以用于单细胞数据,但一些基于单细胞的方法(比如 Pagoda2),效果可能优于前者。
检测细胞组成的变化 Deciphering changes in cell composition
成分分析比较的是不同细胞类型的相对丰度。单变量统计模型,比如泊松回归或是 Wilcoxon 秩和检验可能将部分细胞类型误报为有显著差异,而实际只是样本组成引起的假象。有专门为单细胞设计的 scDC、scCODA 和 tascCODA 测试方法。
推断扰动效应 Inferring perturbation effects
利用细胞培养体系,同时进行大规模多重条件处理细胞的方法称为扰动(perturbations),perturb-seq 或 CROP-seq 等最新技术允许使用多模态数据、全基因组扰动和组合扰动对混池 CRISPR–Cas9 筛选进行分析。
细胞通讯分析 Communication events across cells
细胞通讯分析利用配体、受体及其相互作用的数据库来预测细胞间相互作用。推荐使用 LIANA,它提供了多种方法和数据库。此外还有CellChat、CellPhoneDB、SingleCellSignalR;提供细胞内活动的补充估计的 Nichenet 和 Cytotalk。
原文
原文中还有关于单细胞 ATAC、单细胞蛋白组、空间转录组等内容,但目前还不太关注,就先不记录了。如果有需要还请阅读原文。